Mukopolysacharidosa
Je velmi obtížné odhalit mukopolysachridózu v její počáteční fázi. Dítě je při narození zcela v pořádku a zpočátku se vyvíjí v mezích normy. Obtíže nastávají podle typu obvykle po prvním roce života a pozvolna se zhoršují.
Dítě se začne zastavovat v psychomotorickém vývoji, stává se velmi neklidným, ztrácí potřebu spánku, je inkontinentní a postupně u něho dojde k úbytku všech rozumových schopností. Průběh onemocnění se liší u jednotlivých typů mukopolysacharidóz a některé tkáně nemusí být postiženy vůbec. V současné době neexistuje žádný lék, který by nemoc zcela vyléčil. U některých typů lze průběh zastavit nebo alespoň zpomalit.
Mohu poznat, že mé dítě onemocní?
Mezi lidmi se šíří řada mýtů, více či méně podivných, které přičítají existenci vrozeného onemocnění „vadě“ rodičů. Najdou se i tací, kteří pronášejí moudra o tom, že „chytří rodiče mají hloupé děti“. Bohužel se objevil i názor lékařky, která jednu z maminek takto nemocného dítěte obvinila, že kdyby v těhotenství nebrala prášky, neměla by takhle nemocné dítě. Zcela ojedinělý výkon předvedla jedna ze sousedek malé Petrušky. Prohlásila, že „ona to má z toho, že ji v zimě nechávali spát venku v kočárku. A tak jim namrzla…“
Jak se to tedy stane? Laicky řečeno – za vším hledej geny. Každý z nás předává dítěti část své „genetické“ výbavy. Každý z nás. Otec i matka. A každý z nás – skutečně každý a nejen rodiče dětí nemocných mukopolysacharidózou – předává svým dětem asi 4 % genů zmutovaných. Změněných, zkrátka jiných. Ovšem to ještě nic neznamená. Nemusí se nic stát. Tak maminka předá „špatný“ gen nějaké nemoci, ovšem tatínek nikoliv. A dítě touto nemocí neonemocní, ale nese tuto „zmutovanou“ informaci dál. Nemocí jsou desítky a stovky. U mukopolysacharidózy je to obdobné. S tím rozdílem, že je o mnoho vzácnější.
V podstatě lze říci, že osud (Bůh, příroda) s námi všemi hraje zvláštní ruletu. Kolo se točí a kulička téhle zvláštní hry klouže na okraji hracího pole jménem život… Černá, červená – sudá, lichá. Ruleta osudu se točí dál. Až se u někoho se zastaví v políčku s názvem – mukopolysacharidóza. A najednou – náhodou a statisticky velmi nepravděpodobně – se potká s úplně stejně změněným genem partnera. A tak v podstatě všichni, znovu zdůrazněme všichni, hrajeme tuto ruletu. Jenom mnozí o jejích pravidlech nevědí. A proto mohou – nevědomě pravit – nám se to nemůže stát…
Rozdělení mukopolysacharidózy
podle druhu chybějícího enzymu
Mukolipidózy a jiná střádavá onemocnění
Následující výčet představuje nemoci, které jsou mukopolysacharidózám velmi podobné:
-
ML II
-
ML III
-
ML IV
-
Siallidóza (dříve ML I)
-
Fukosidóza
-
Alfa Mannosidóza
-
Aspartylglukosaminurie
-
Neuronální ceroidlipofuscinóza
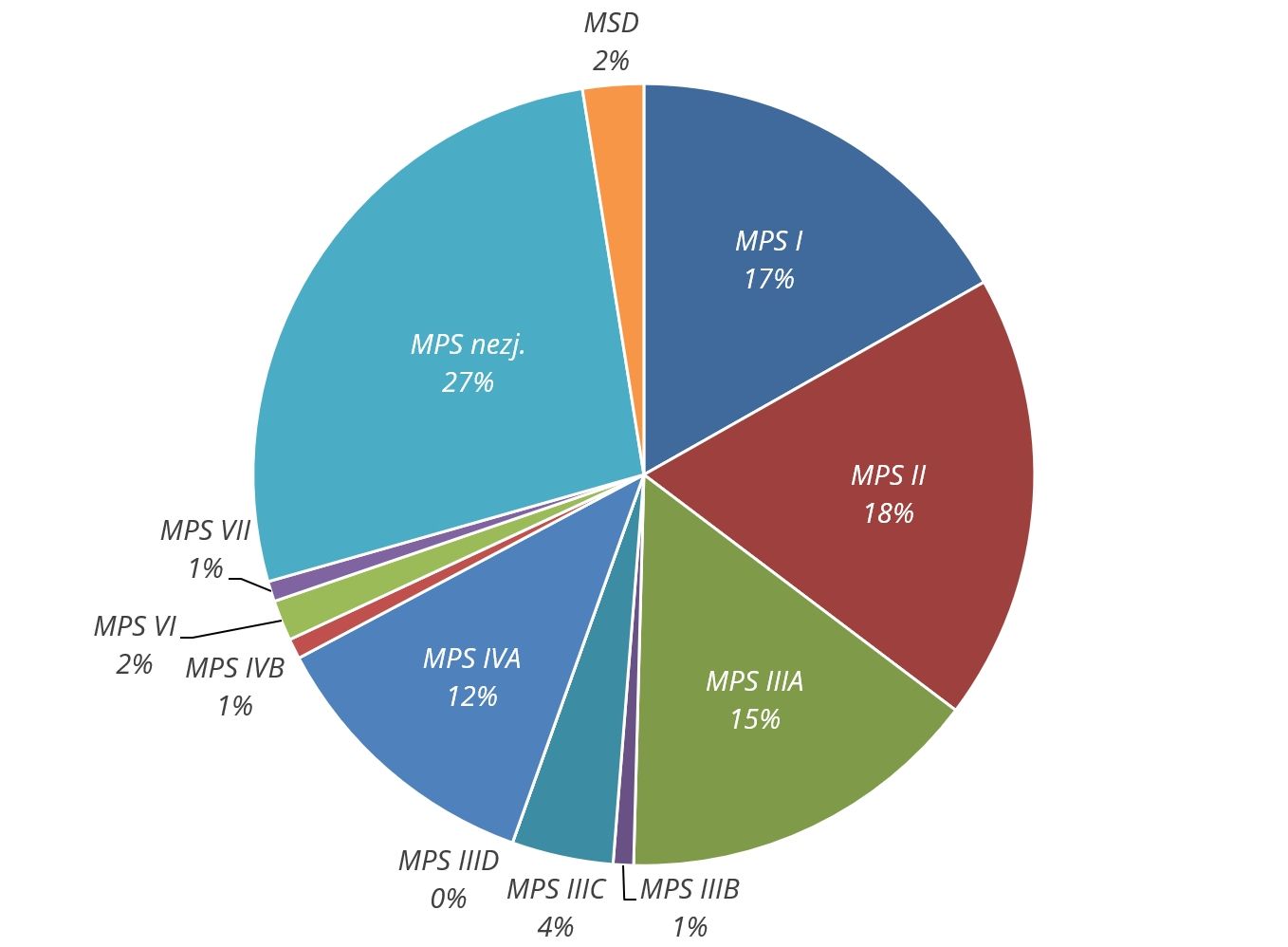
V tabulce a grafu jsou uvedeny počty pacientů s různými typy mukopolysacharidóz, kteří byli diagnostikováni v České republice v letech 1975–2008. (z publikace Poupětová a spol., JIMD 2010)
| Typ MPS | Počet pacientů |
|---|---|
MPS I | 20 |
MPS II | 22 |
MPS IIIA | 18 |
MPS IIIB | 1 |
MPS IIIC | 5 |
MPS IIID | 0 |
MPS IVA | 14 |
MPS IVB | 1 |
MPS VI | 2 |
MPS VII | 1 |
MPS nezj. | 32 |
MSD | 3 |
celkem | 119 |
Kudy se ubírá léčba a její výzkum?
Vědecké týmy v celé řadě zemí intenzivně pracují na několika způsobech léčby:
Transplantace kmenových buněk
Transplantace kmenových buněk kostní dřeně nebo pupečníkové krve je v současné době jedna z forem léčby, avšak za přesně vymezených podmínek. Lze ji aplikovat pouze u některých typů MPS, u kterých bylo prokázáno, že je funkční. Věková hranice pro transplantaci bývá obvykle do 2–3 let dítěte. V pozdějším období se výrazně snižuje pravděpodobnost, že transplantované buňky zastaví či alespoň zmírní progresi onemocnění. Transplantace, třebaže její úspěšnost již dosahuje až 80 %, vyžaduje náročnou předtransplantační i potransplantační péči. Nutnou a všeobecně známou podmínkou je, že musí existovat vhodný dárce.
Enzymová terapie
Enzymová terapie je léčba založená na principu dodávání chybějícího enzymu. V současné době už není problém enzym vyrobit, ale vymyslet způsob, jak by ho dokázalo přijmout celé lidské tělo. Ne každý enzym totiž dokáže překonat krevní bariéru mozku a nemůže tak zabránit dalšímu poškozování CNS. Nevýhodou této léčby je skutečnost, že je celoživotní a finančně velmi náročná (cena ročního léčení může dosahovat dle typu a hmotnosti pacienta i více než 10 milionů korun).
Genová terapie
Genová terapie je zatím ve fázi výzkumu. Je založená na principu vnášení „neporušeného“ genu do buněk tkání, které by opravily poškozený genetický kód. Tělo by se tak naučilo produkovat životně důležitý enzym. Genová terapie se zkouší na zvířecích modelech u většiny typů mukopolysacharidóz i u dalších lyzozomálních střádavých onemocnění.
Léčba založená na zamezení přísunu látek do buňky, které způsobují střádání – tzv. substrát redukující.
Tento způsob léčby snižuje tvorbu škodlivých látek, které se u mukopolysacharidóz hromadí v těle v důsledku nefungujícího enzymu. Nyní se aplikuje u pacientů s MPS III.
Včasné stanovení diagnózy onemocnění v rodině přináší u některých typů možnost správné a včasné léčby, která může zmírnit, nebo dokonce zastavit další projevy choroby. Pro potenciální další potomky v rodině je následně k dispozici prenatální diagnostika. Ta dovede stanovit, zda plod je, či není postižen. V případě postižení plodu mohou rodiče, podle jejich rozhodnutí, těhotenství včas ukončit.
Kontakt
prof. Mgr. PedDr. Jan Michalík, Ph.D.
předseda Společnosti
+420 777 809 040
spmps@seznam.cz
Mgr. Petra Hájková, Ph.D.
krizová interventka, asistentka Společnosti
+420 733 690 750
Bc. Martina Vysloužilová Dis.
PR a média
+420 603 359 126
Fakturační údaje
Společnost pro mukopolysacharidosu, z. s.
Chaloupky 4/35, Týneček
779 00 Olomouc
IČ: 619 85 112
Číslo účtu: 108775975/0300
Partnerství
Staňte se partnerem Společnosti pro mukopolysacharidosu, podpořte rodiny s nemocnými dětmi a darujte nemocným dětem příležitost žít a zemřít doma.